タグ: algorithm chemistry variational
このチュートリアルでは、水素分子(H₂)の基底状態エネルギーを求めるための変分量子固有値ソルバー(VQE)アルゴリズムの実装について解説します。分子ハミルトニアンの生成には OpenFermion を使用します。
ワークフローは以下の通りです:
分子ハミルトニアンを量子ビット演算子へ変換
パラメータ化された量子回路(アンザッツ)の作成
VQEによる最適化の実装
原子間距離ごとのエネルギー地形の解析
量子コンピューティングを用いた量子化学問題の解法を紹介し、特にH₂分子の最小エネルギー構造の探索に焦点を当てます。
# 必要なパッケージは以下のコマンドでインストールできます
# !pip install openfermion pyscf openfermionpyscfimport os
import warnings
import matplotlib.pyplot as plt
import numpy as np
import openfermion.chem as of_chem
import openfermion.transforms as of_trans
import openfermionpyscf as of_pyscf
from qiskit_aer.primitives import EstimatorV2
from scipy.optimize import minimize
import qamomile.circuit as qmc
import qamomile.observable as qm_o
from qamomile.circuit.algorithm.basic import cx_entangling_layer, ry_layer, rz_layer
from qamomile.qiskit import QiskitTranspiler
from qamomile.qiskit.transpiler import QiskitExecutor
docs_test_mode = os.environ.get("QAMOMILE_DOCS_TEST") == "1"水素分子のハミルトニアンの作成¶
basis = "sto-3g"
multiplicity = 1
charge = 0
distance = 0.977
geometry = [["H", [0, 0, 0]], ["H", [0, 0, distance]]]
description = "tmp"
molecule = of_chem.MolecularData(geometry, basis, multiplicity, charge, description)
molecule = of_pyscf.run_pyscf(molecule, run_scf=True, run_fci=True)
n_qubit = molecule.n_qubits
n_electron = molecule.n_electrons
# H2 を STO-3G 基底で扱うと、空間軌道 2 個 -> スピン軌道 4 個 -> 4 量子ビット、
# 電子数は 2。
assert n_qubit == 4
assert n_electron == 2
fermionic_hamiltonian = of_trans.get_fermion_operator(
molecule.get_molecular_hamiltonian()
)
jw_hamiltonian = of_trans.jordan_wigner(fermionic_hamiltonian)Qamomile ハミルトニアンへの変換¶
このセクションでは、OpenFermionのハミルトニアンをQamomileフォーマットに変換します。Jordan-Wigner変換を適用してフェルミ粒子演算子を量子ビット演算子へ変換し、その後、カスタム変換関数を用いてQamomileに適したハミルトニアン表現を作成します。
def operator_to_qamomile(operators: tuple[tuple[int, str], ...]) -> qm_o.Hamiltonian:
pauli = {"X": qm_o.X, "Y": qm_o.Y, "Z": qm_o.Z}
H = qm_o.Hamiltonian()
H.constant = 1.0
for ope in operators:
H *= pauli[ope[1]](ope[0])
return H
def openfermion_to_qamomile(of_h) -> qm_o.Hamiltonian:
H = qm_o.Hamiltonian()
for k, v in of_h.terms.items():
if len(k) == 0:
H.constant += v
else:
H += operator_to_qamomile(k) * v
return H
hamiltonian = openfermion_to_qamomile(jw_hamiltonian)
assert hamiltonian.num_qubits == n_qubitVQE アンザッツの作成¶
このセクションでは、VQEアルゴリズムのための Hardware Efficient SU(2) アンザッツを @qkernel デコレータを用いて作成します。アンザッツとは、試行波動関数を準備するパラメータ付き量子回路です。ry_layer、rz_layer および線形 CX エンタングル層を組み合わせて構築し、最後に expval でハミルトニアンの期待値を計算します。
@qmc.qkernel
def vqe_ansatz(
n: qmc.UInt,
reps: qmc.UInt,
thetas: qmc.Vector[qmc.Float],
H: qmc.Observable,
) -> qmc.Float:
q = qmc.qubit_array(n, name="q")
for r in qmc.range(reps):
base = r * 2 * n
q = ry_layer(q, thetas, base)
q = rz_layer(q, thetas, base + n)
q = cx_entangling_layer(q)
# Final rotation layer
final_base = reps * 2 * n
q = ry_layer(q, thetas, final_base)
q = rz_layer(q, thetas, final_base + n)
return qmc.expval(q, H)Qiskitを用いたVQEの実行¶
このセクションでは、QiskitTranspiler を使って VQE カーネルを実行可能オブジェクトにトランスパイルします。デフォルトの executor がこのオブジェクトを実行し、qkernel で定義した expval による期待値を返します。そのため、ユーザーは最適化ループのみ実装すれば問題ありません。
transpiler = QiskitTranspiler()
reps = 4
executable = transpiler.transpile(
vqe_ansatz,
bindings={"n": n_qubit, "reps": reps, "H": hamiltonian},
parameters=["thetas"],
)
# Transpiled quantum circuit
executable.quantum_circuit.draw("mpl")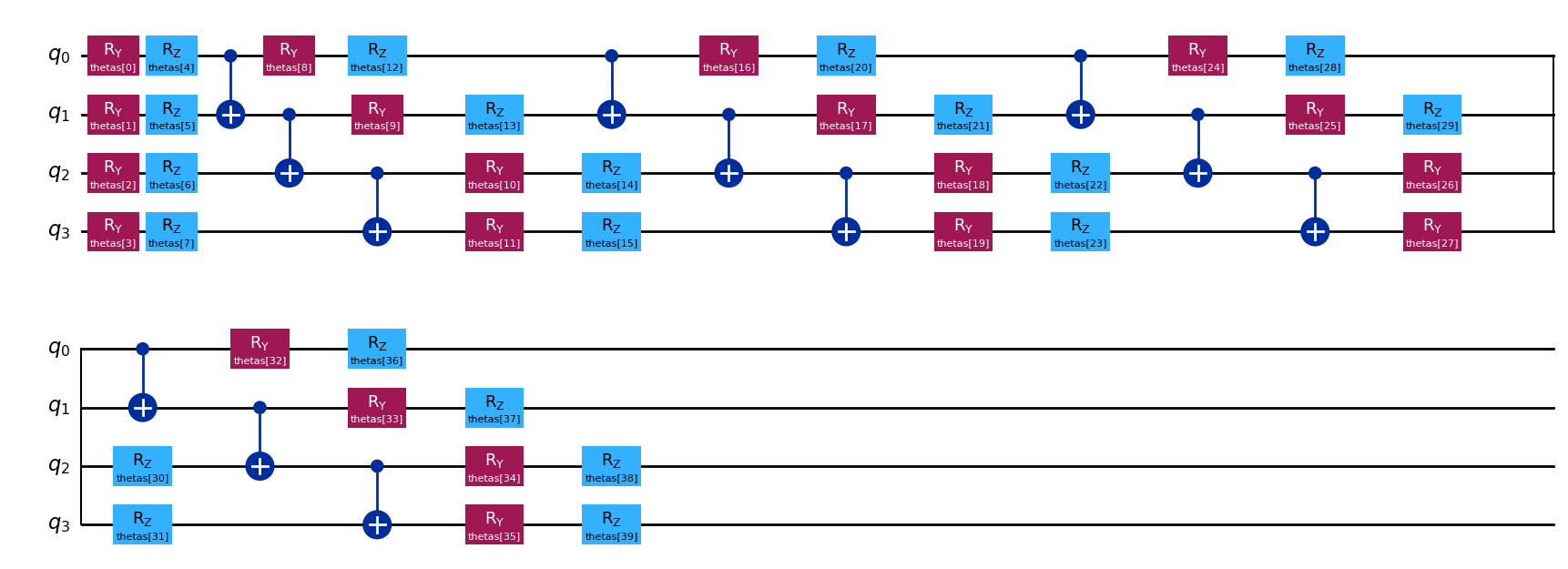
cost_history = []
executor = QiskitExecutor(estimator=EstimatorV2())
def cost_fn(param_values):
job = executable.run(executor, bindings={"thetas": list(param_values)})
return job.result()
def cost_callback(param_values):
cost_history.append(cost_fn(param_values))
num_params = len(executable.parameter_names)
rng = np.random.default_rng(42)
initial_params = rng.uniform(0, np.pi, num_params)
# 各 rep は RY 層 (n パラメータ) と RZ 層 (n パラメータ) を、最後の追加層も
# RY + RZ ペアをもう 1 つ出すので、パラメータ数は (reps + 1) * 2 * n_qubit。
assert num_params == (reps + 1) * 2 * n_qubit
assert initial_params.shape == (num_params,)
# VQE 最適化を実行します
maxiter = 1 if docs_test_mode else 50
warnings.filterwarnings("ignore", message="Maximum number of iterations")
result = minimize(
cost_fn,
initial_params,
method="BFGS",
options={"disp": True, "maxiter": maxiter, "gtol": 1e-6},
callback=cost_callback,
)
print(result)
# 変分原理: BFGS の予算が短くても、試行エネルギーは FCI 基底エネルギーの上界。
# 上で ``run_fci=True`` を渡しているので ``molecule.fci_energy`` は埋まっている。
# openfermion の stub は ``run_fci=False`` のケースも考慮して ``float | None``
# と宣言しているので、ここで narrow する。
fci_ref = molecule.fci_energy
assert fci_ref is not None
assert result.fun >= fci_ref - 1e-9
assert len(result.x) == num_params Current function value: -1.105860
Iterations: 50
Function evaluations: 2091
Gradient evaluations: 51
message: Maximum number of iterations has been exceeded.
success: False
status: 1
fun: -1.105859672342776
x: [ 3.123e+00 1.022e+00 ... 1.306e+00 2.499e+00]
nit: 50
jac: [-2.432e-04 7.428e-05 ... -2.052e-04 -1.994e-04]
hess_inv: [[ 3.779e+00 5.352e-01 ... 5.601e-01 4.337e-01]
[ 5.352e-01 1.988e+00 ... -7.801e-01 -2.818e-01]
...
[ 5.601e-01 -7.801e-01 ... 3.531e+00 2.205e+00]
[ 4.337e-01 -2.818e-01 ... 2.205e+00 3.404e+00]]
nfev: 2091
njev: 51
plt.plot(cost_history)
plt.plot(
range(len(cost_history)),
[fci_ref] * len(cost_history),
linestyle="dashed",
color="black",
label="Exact Solution",
)
plt.legend()
plt.show()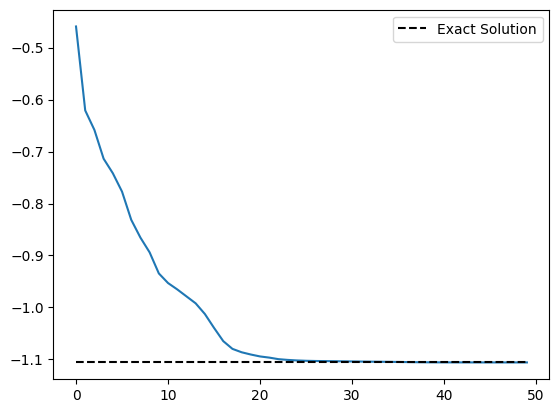
原子同士の距離を変更する¶
def hydrogen_molecule(bond_length):
basis = "sto-3g"
multiplicity = 1
charge = 0
geometry = [["H", [0, 0, 0]], ["H", [0, 0, bond_length]]]
description = "tmp"
molecule = of_chem.MolecularData(geometry, basis, multiplicity, charge, description)
molecule = of_pyscf.run_pyscf(molecule, run_scf=True, run_fci=True)
fermionic_hamiltonian = of_trans.get_fermion_operator(
molecule.get_molecular_hamiltonian()
)
jw_hamiltonian = of_trans.jordan_wigner(fermionic_hamiltonian)
return openfermion_to_qamomile(jw_hamiltonian), molecule.fci_energy
n_points = 3 if docs_test_mode else 15
bond_lengths = np.linspace(0.2, 1.5, n_points)
assert bond_lengths.shape == (n_points,)
energies = []
for bond_length in bond_lengths:
hamiltonian, fci_energy = hydrogen_molecule(bond_length)
# 結合長を変えても H2 は 4 量子ビット問題のまま。
assert hamiltonian.num_qubits == 4
executable = transpiler.transpile(
vqe_ansatz,
bindings={"n": hamiltonian.num_qubits, "reps": reps, "H": hamiltonian},
parameters=["thetas"],
)
num_params = len(executable.parameter_names)
initial_params = rng.uniform(0, np.pi, num_params)
result = minimize(
cost_fn,
initial_params,
method="BFGS",
options={"maxiter": maxiter, "gtol": 1e-6},
)
energies.append(result.fun)
# ``run_fci=True`` を渡しているので ``fci_energy`` は populate されている。
# openfermion stub の ``float | None`` をここで narrow。
assert fci_energy is not None
# 変分原理はどの結合長でも成り立つ。
assert result.fun >= fci_energy - 1e-9
print("distance: ", bond_length, "energy: ", result.fun, "fci_energy: ", fci_energy)
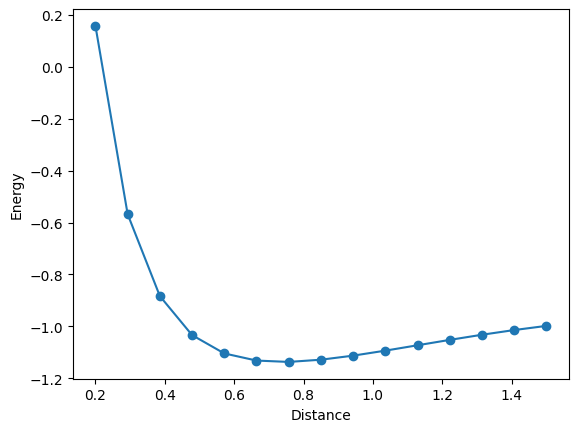
assert len(energies) == n_pointsdistance: 0.2 energy: 0.15749329365461995 fci_energy: 0.15748213479836526
distance: 0.29285714285714287 energy: -0.5679193889010842 fci_energy: -0.5679447209710013
distance: 0.38571428571428573 energy: -0.8833483653566578 fci_energy: -0.8833596636183383
distance: 0.4785714285714286 energy: -1.033582342419856 fci_energy: -1.0336011797110967
distance: 0.5714285714285714 energy: -1.1041774579732506 fci_energy: -1.1042094222435166
distance: 0.6642857142857144 energy: -1.1316536828860908 fci_energy: -1.1323508827075506
distance: 0.7571428571428571 energy: -1.136896895687888 fci_energy: -1.1369026717971333
distance: 0.8500000000000001 energy: -1.128359326275411 fci_energy: -1.128361878458112
distance: 0.9428571428571428 energy: -1.1127239395799284 fci_energy: -1.1127252078468768
distance: 1.0357142857142858 energy: -1.093476087896891 fci_energy: -1.093476088229404
distance: 1.1285714285714286 energy: -1.0727482357543656 fci_energy: -1.0727578805453502
distance: 1.2214285714285713 energy: -1.0520071096366461 fci_energy: -1.052008162170845
distance: 1.3142857142857143 energy: -1.0322163897827572 fci_energy: -1.0322400306247084
distance: 1.4071428571428573 energy: -1.014138609349133 fci_energy: -1.0141470586695496
distance: 1.5 energy: -0.998088727242389 fci_energy: -0.9981493534714101
plt.plot(bond_lengths, energies, "-o")
plt.xlabel("Distance")
plt.ylabel("Energy")
plt.show()